In short, a complete workflow from Illumina reads to genome assembly, through annotation and comparative genomics.
This example uses just the Illumina data (https://www.ebi.ac.uk/ena/browser/view/ERR3335404) generated for the study “Whole genome sequence of Mycobacterium ulcerans CSURP7741, a French Guyana clinical isolate” (https://www.ebi.ac.uk/ena/browser/view/PRJEB30628), “P7741” as a short identity code. Mycobacterium ulcerans is an environmental non-tuberculous mycobacterium responsible for Buruli ulcer.
One reference genome paper is “Complete genome sequence of the frog pathogen Mycobacterium ulcerans ecovar Liflandii” and it’s sequence https://www.ncbi.nlm.nih.gov/nuccore/NC_020133. Mycobacterium ulcerans liflandii 128FXT, complete sequence has NCBI Reference Sequence: NC_020133.1 and is refered to as just “Liflandii” (sequence stored as ./genomes/Liflandii.fasta). Other reference genomes of Mycobacterium ulcerans are utilized in the workflow (Agy99, SGL03 and Shinsuense) and one of Mycobacterium tuberculosis (H37Rv). This work is based upon the excellent presentations and analysis demonstrated by Vincent Appiah (vincentappiah, link) and Daren Ginete (link). The work has been reproduced, adapted and extended in order that it works on a Mac laptop with updated versions of analysis software available in February 2024 and using the Quarto package for presentation and re-usability.
# use clean install of conda on machine in private space source ~/anaconda3/etc/profile.d/conda.shconda-V# all those envs explained on Set Up pageconda env listconda activate bacterial-genomics-tutorial-sw7# uncomment appropriately to re-run# download data to data # chmod +x download_data.sh# ./download_data.sh# run fastqc# mkdir QC_RAW_READS# fastqc data/*.fastq.gz -o QC_RAW_READS# show the raw datals-lh data/ |perl-nae'print "@F[4..8]\n"'conda deactivate
conda 24.1.2
# conda environments:
#
base * /Users/sw/anaconda3
bacterial-genomics-tutorial-sw1 /Users/sw/anaconda3/envs/bacterial-genomics-tutorial-sw1
bacterial-genomics-tutorial-sw2 /Users/sw/anaconda3/envs/bacterial-genomics-tutorial-sw2
bacterial-genomics-tutorial-sw3 /Users/sw/anaconda3/envs/bacterial-genomics-tutorial-sw3
bacterial-genomics-tutorial-sw4 /Users/sw/anaconda3/envs/bacterial-genomics-tutorial-sw4
bacterial-genomics-tutorial-sw5 /Users/sw/anaconda3/envs/bacterial-genomics-tutorial-sw5
bacterial-genomics-tutorial-sw7 /Users/sw/anaconda3/envs/bacterial-genomics-tutorial-sw7
example /Users/sw/anaconda3/envs/example
for_drep /Users/sw/anaconda3/envs/for_drep
just_quast /Users/sw/anaconda3/envs/just_quast
40M Feb 23 16:02 P7741_R1.fastq.gz
45M Feb 23 16:02 P7741_R2.fastq.gz
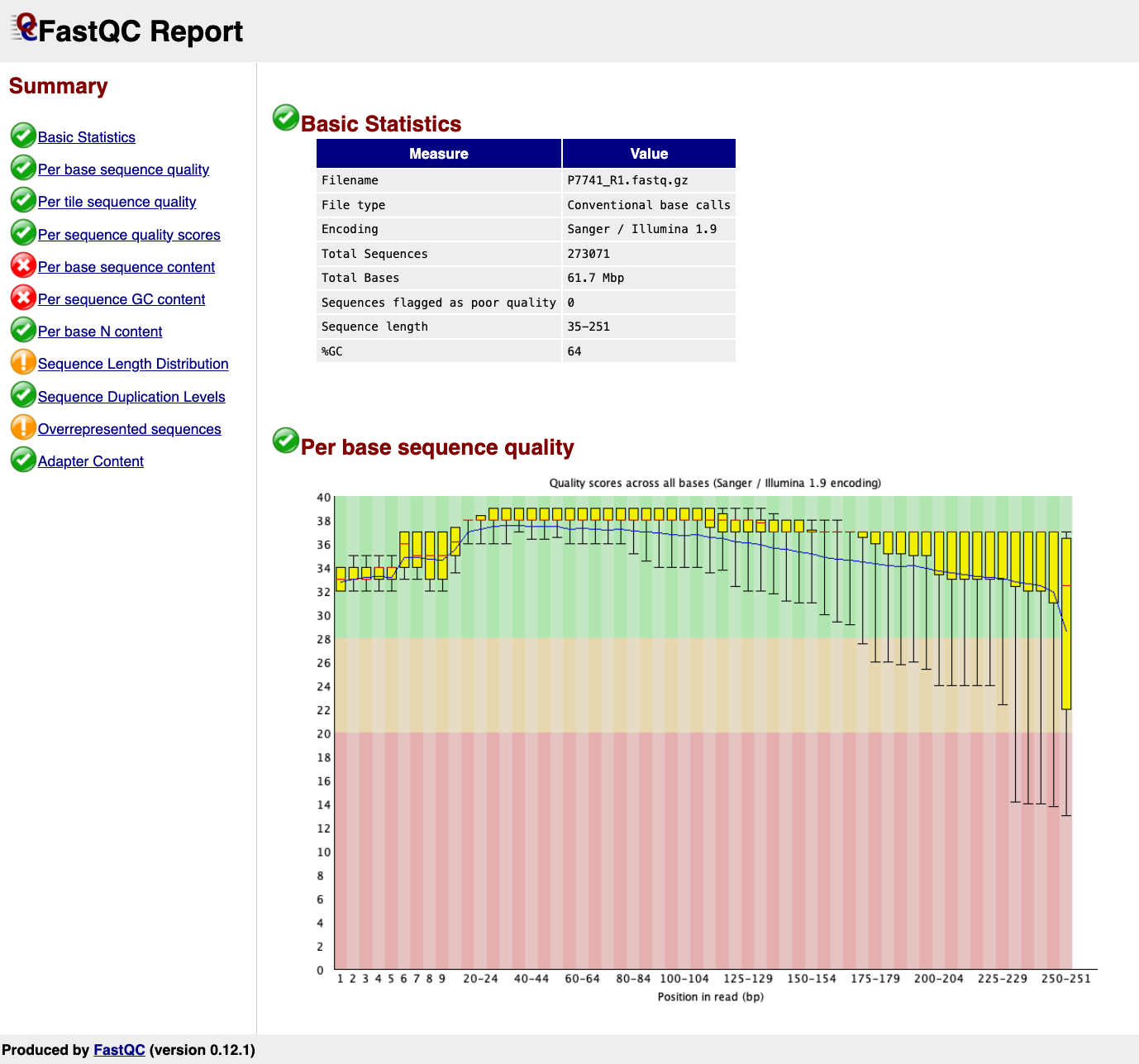
Results files are QC html reports. vincentappiah’s results are different around 16:29. In video he speaks of sickle trimming, but the code uses trimmomatic.
# Real problems setting up spades with conda - notes on Set Up page# Solution was to get the spades Mac binaries## It runs but takes quite some time on my mini-server# Therefore delete directory# P7741_SPADES_OUT# and re-run if needed# ~/SPAdes-3.15.5-Darwin/bin/spades.py --careful -o P7741_SPADES_OUT -1 trimmed_reads/P7741_R1.fastq.gz -2 trimmed_reads/P7741_R2.fastq.gzecho"number of contigs"grep'>' P7741_SPADES_OUT/contigs.fasta |wc-lecho""echo"number of scaffolds"grep'>' P7741_SPADES_OUT/scaffolds.fasta |wc-lecho""echo"directory listing"ls-1 P7741_SPADES_OUT
number of contigs
3243
number of scaffolds
3232
directory listing
K21
K33
K55
K77
assembly_graph.fastg
assembly_graph_after_simplification.gfa
assembly_graph_with_scaffolds.gfa
before_rr.fasta
contigs.fasta
contigs.paths
corrected
dataset.info
input_dataset.yaml
misc
mismatch_corrector
params.txt
pipeline_state
run_spades.sh
run_spades.yaml
scaffolds.fasta
scaffolds.paths
spades.log
tmp
warnings.log
Polishing
with
bwa (alignment via Burrows-Wheeler transformation)
pilon (automatically improve draft assemblies and find variation among strains including large event detection)
Use script to do 4 rounds (not guaranteed to be optimal) of polishing. Runs but takes quite some time on my mini-server but it goes.
# conda source ~/anaconda3/etc/profile.d/conda.shconda activate bacterial-genomics-tutorial-sw7# To clean re-run delete dir polishing_process# re-run script# ./polish.sh# list out the files developed find polishing_processconda deactivate
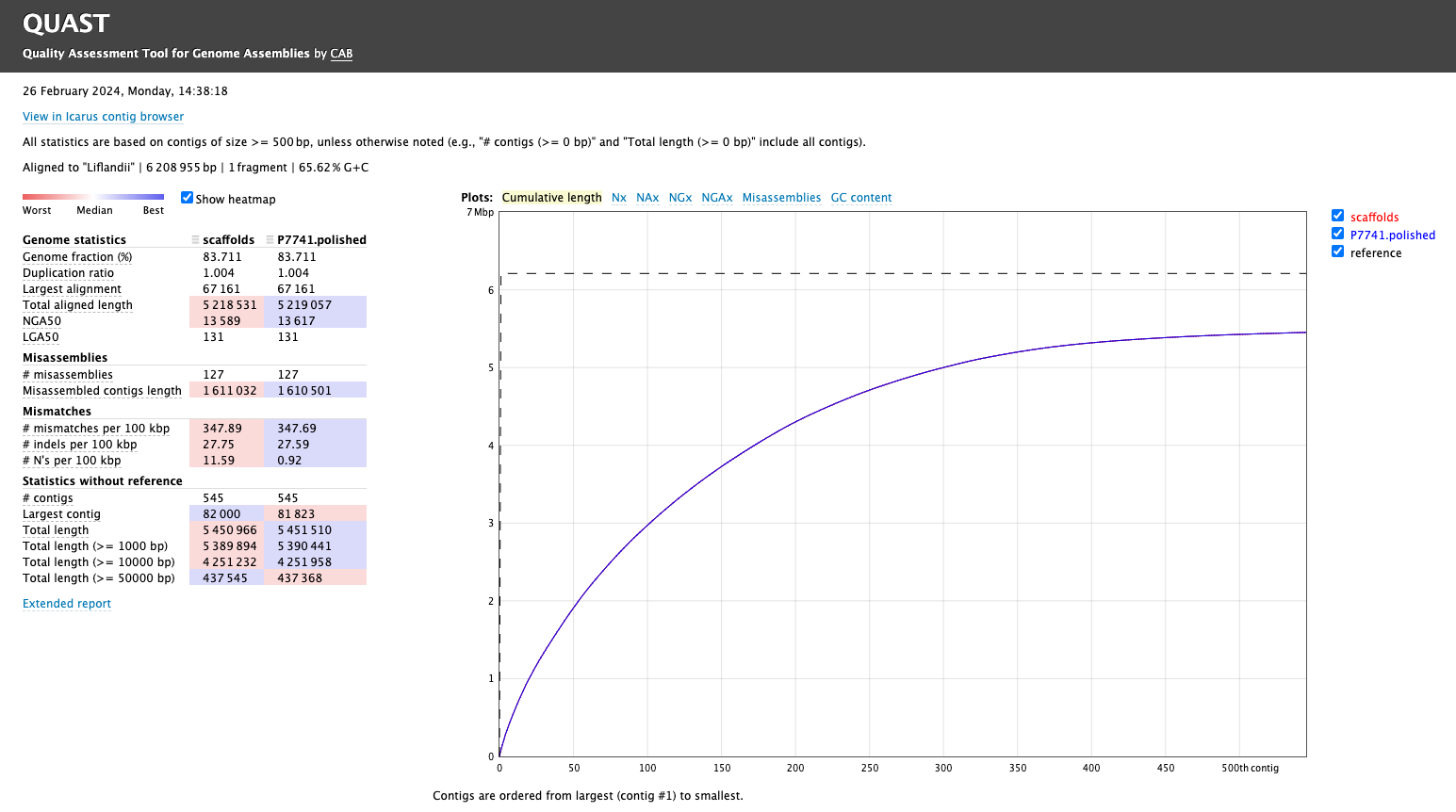
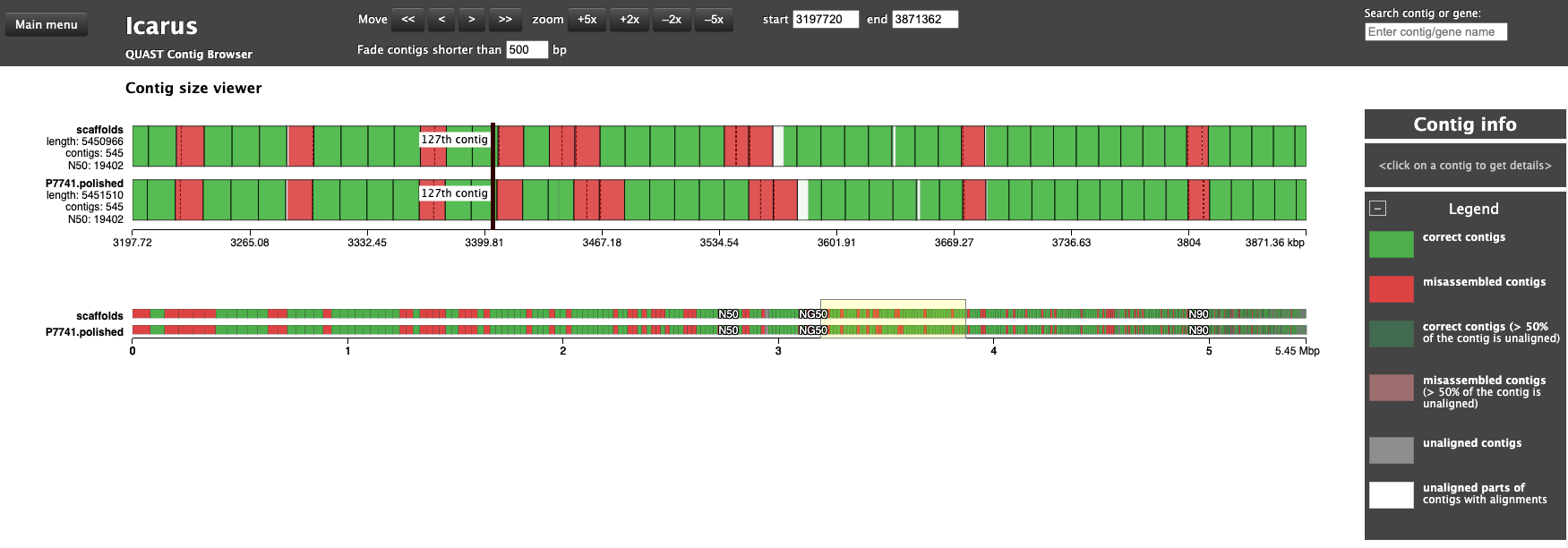
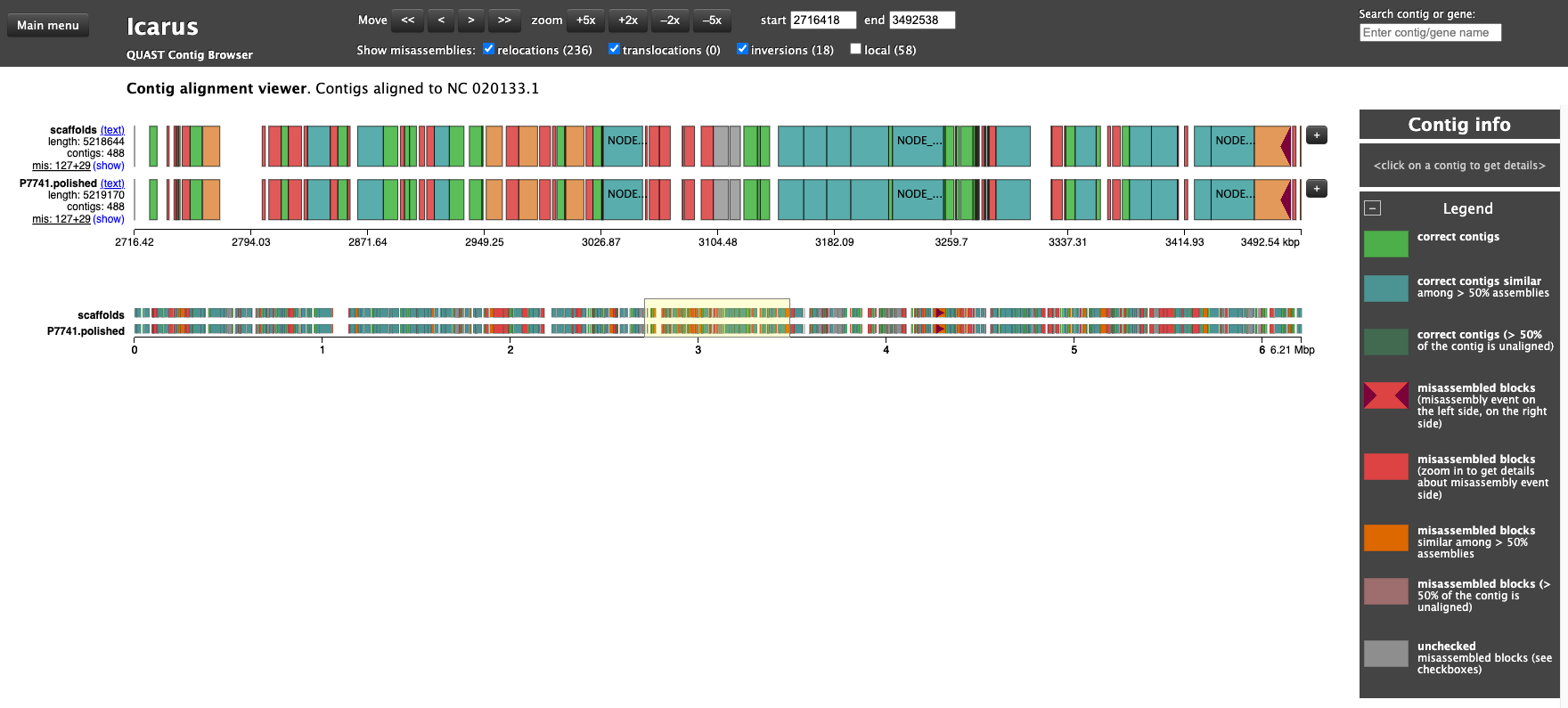
# condasource ~/anaconda3/etc/profile.d/conda.sh# env just with the quast softwareconda activate just_quast# genomes dir taken from vincentappiah repo# quast throws an error with default script# -t 1 solves # other potential issue may exist as the install of quast was not completed...# see SetUp page# uncomment to run again# mkdir QC_ASSEMBLY# quast.py -t 1 -o QC_ASSEMBLY -R genomes/Liflandii.fasta P7741_SPADES_OUT/scaffolds.fasta P7741.polished.fasta# Results notes# N50 quality didn't change with polishing, GC% didn't change,# Slight change in misassemblies # list out the top dir ls-1 QC_ASSEMBLYconda deactivate
# conda source ~/anaconda3/etc/profile.d/conda.shconda activate bacterial-genomics-tutorial-sw7# genomes/Agy99.fasta is NC_008611.1 Mycobacterium ulcerans Agy99, complete sequence# note this is a different genome to genomes/Liflandii.fastaragtag.py-c# uncomment to re-run## ragtag.py scaffold genomes/Agy99.fasta P7741.polished.fasta -o P7741_reordered# Extract the reordered contig with a custom (modified!) python script# The scripts accept name of the ragtag file containing the reordered contigs # and accession number for the reference genomepython extract_reordered.py P7741_reordered/ragtag.scaffold.fasta NC_008611.1# at this stage vincentappiah gets length of 5291728 # and a gc of 64.98# potentially the difference is due to newer version of ragtag# i.e. v1.0.2 vs RagTag v2.1.0conda deactivate
Alonge, Michael, et al. "RaGOO: fast and accurate reference-guided scaffolding of draft genomes."
Genome biology 20.1 (2019): 1-17.
https://doi.org/10.1186/s13059-019-1829-6
GC Percent, opt=ignore: 65.073454
GC Percent, opt=remove: 65.701833
GC Percent, opt=weighted: 65.551660
Sequence Length: 5012494 bp
draft genome sequence extracted
Multi-Locus Sequence Typing (MLST) and Antibiotic Resistance
The results show the expected identity (7 alleles of mycobacteria) and there is an antibiotic resistance gene.
But need to use cgMLST, wgMLST and more…
Need to look at “chewBBACA is a comprehensive pipeline including a set of functions for the creation and validation of whole genome and core genome MultiLocus Sequence Typing (wg/cgMLST) schemas..”
Alleles
[11:44:55] This is mlst 2.23.0 running on darwin with Perl 5.030003
[11:44:55] Checking mlst dependencies:
[11:44:55] Found 'blastn' => /Users/sw/anaconda3/envs/bacterial-genomics-tutorial-sw7/bin/blastn
[11:44:55] Found 'any2fasta' => /Users/sw/anaconda3/envs/bacterial-genomics-tutorial-sw7/bin/any2fasta
[11:44:55] Found blastn: 2.15.0+ (002015)
[11:44:55] Excluding 3 schemes: ecoli abaumannii vcholerae_2
[11:44:57] Found exact allele match mycobacteria_2.S7-10
[11:44:57] Found exact allele match mycobacteria_2.L16-450
[11:44:57] Found exact allele match mycobacteria_2.S12-394
[11:44:57] Found exact allele match mycobacteria_2.L19-11
[11:44:57] Found exact allele match mycobacteria_2.S19-13
[11:44:57] Found exact allele match mycobacteria_2.L35-10
[11:44:57] Found exact allele match mycobacteria_2.S14Z-10
[11:44:57] mlst also supports --json output for the modern bioinformatician.
[11:44:57] Done.
Antibiotic resistance
Using nucl database ncbi: 5386 sequences - 2023-Nov-4
Processing: P7741.reordered.fasta
Found 1 genes in P7741.reordered.fasta
Tip: you can use the --summary option to combine reports in a presence/absence matrix.
Done.
#FILE SEQUENCE START END STRAND GENE COVERAGE COVERAGE_MAP GAPS %COVERAGE %IDENTITY DATABASE ACCESSION PRODUCT RESISTANCE
P7741.reordered.fasta P7741 1110389 1110934 - aac(2')-Ic 1-546/546 ========/====== 2/2 99.82 82.27 ncbi NG_047229.1 aminoglycoside N-acetyltransferase AAC(2')-Ic GENTAMICIN;TOBRAMCYIN
# conda source ~/anaconda3/etc/profile.d/conda.shconda activate bacterial-genomics-tutorial-sw7cpus=4# uncomment to re-run# ran is around 10 minutes with 4 cores#prokka --cpus $cpus --kingdom Bacteria --locustag P7741 --outdir P7741_annotation --prefix P7741 --addgenes P7741.reordered.fasta# statsecho"Summary"cat P7741_annotation/P7741.txt echo""# get some counts of genome features# vincentappiah numbers differ for reason speculated over in above sectionsecho"Another bespoke summary"python get_annot_stats.py P7741_annotation P7741echo""# show first 3 protein sequencesecho"First 3 protein sequences"head-13 P7741_annotation/P7741.faaecho""# show some 3 genes/CDSecho"First 3 genes / CDS"head-7 P7741_annotation/P7741.tsvecho""# show some pseudogenesecho"Some pseudogenes"./get_pseudo.pl P7741_annotation/P7741.faa > P7741_annotation/P7741.pseudo.txthead-10 P7741_annotation/P7741.pseudo.txtecho"...."conda deactivate
FastANI is developed for fast alignment-free computation of whole-genome Average Nucleotide Identity (ANI).
Script dendrogram.sh calls dRep: it is installed in conda bacterial-genomics-tutorial-sw7 but not possible to add fastani to this build do to errors (with perl versions if I remember correctly), so build a particular environment for dRep (see Set Up page).
# conda source ~/anaconda3/etc/profile.d/conda.sh# particular environment for dRepconda activate for_drep# dRep check_dependencies# uncomment to re-run# ./dendogram.shconda deactivate
Some of the outputs from the PDFs generated are given as screen-shot images here.
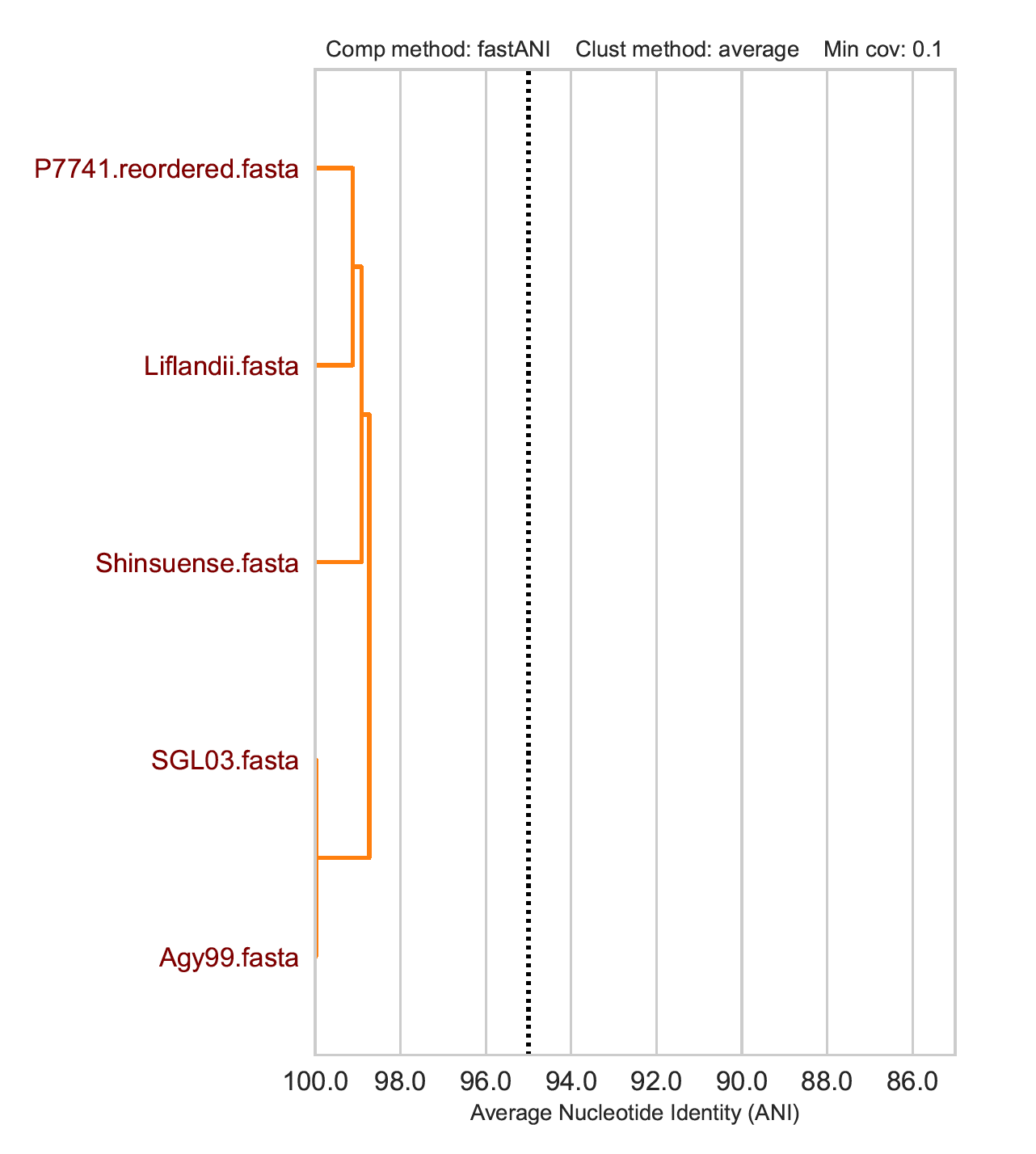
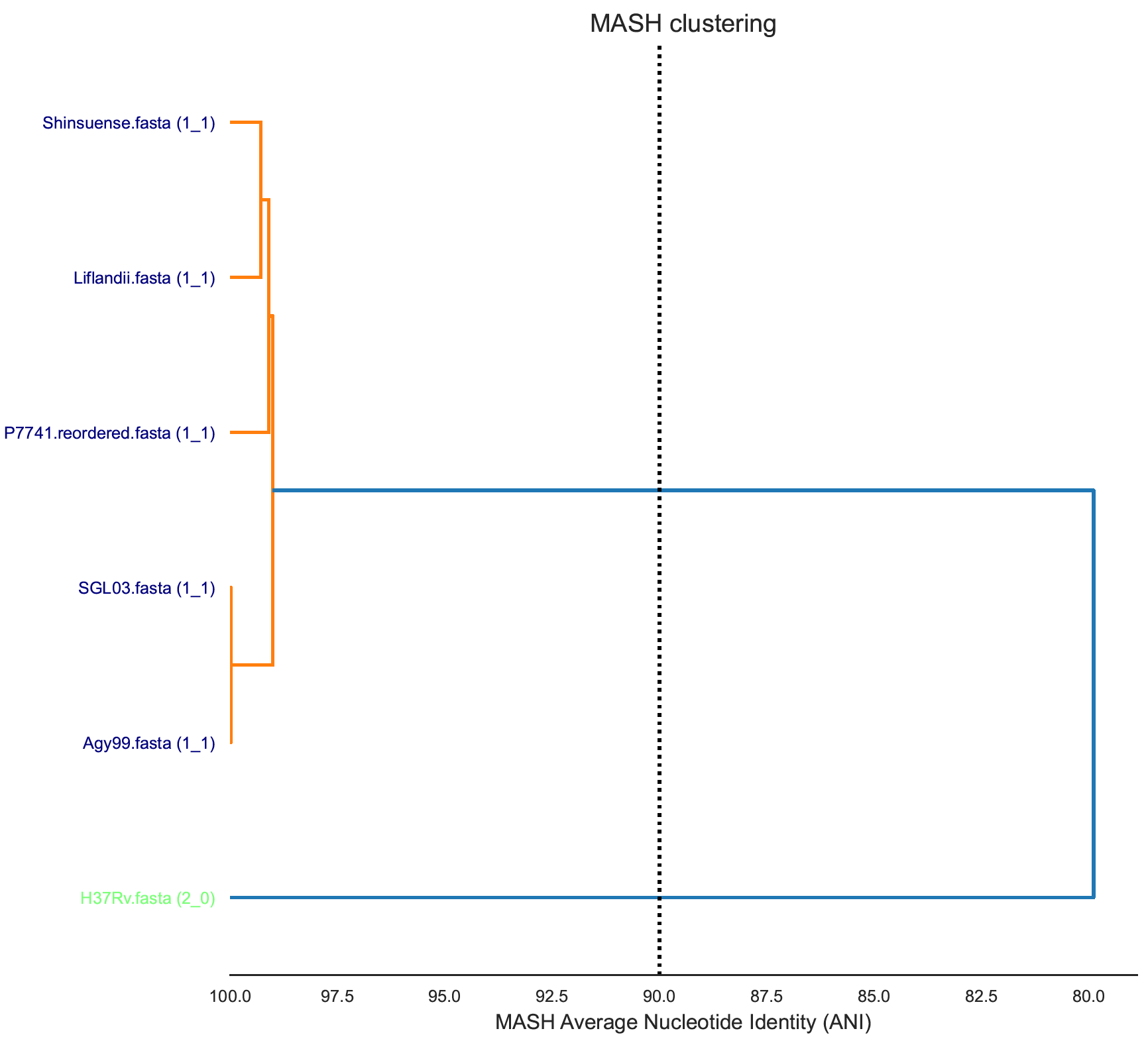
Cluster of P7741/Liflandii/Shinsuense is different between methods ANI methods but agreement with the distinction of SGL03 and Agy99 that are approaching 100% Average Nucleotide Identity.
fastANI cluster: just the Mycobacterium ulcerans genomes
MASH cluster: H37Rv (green label - hard to read in image) is Mycobacterium tuberculosis and is clearly very different, an outlier with respect to ANI, to the Mycobacterium ulcerans genomes.
Roary is a high speed stand alone pan genome pipeline, which takes annotated assemblies in GFF3 format (produced by Prokka (Seemann, 2014)) and calculates the pan genome.
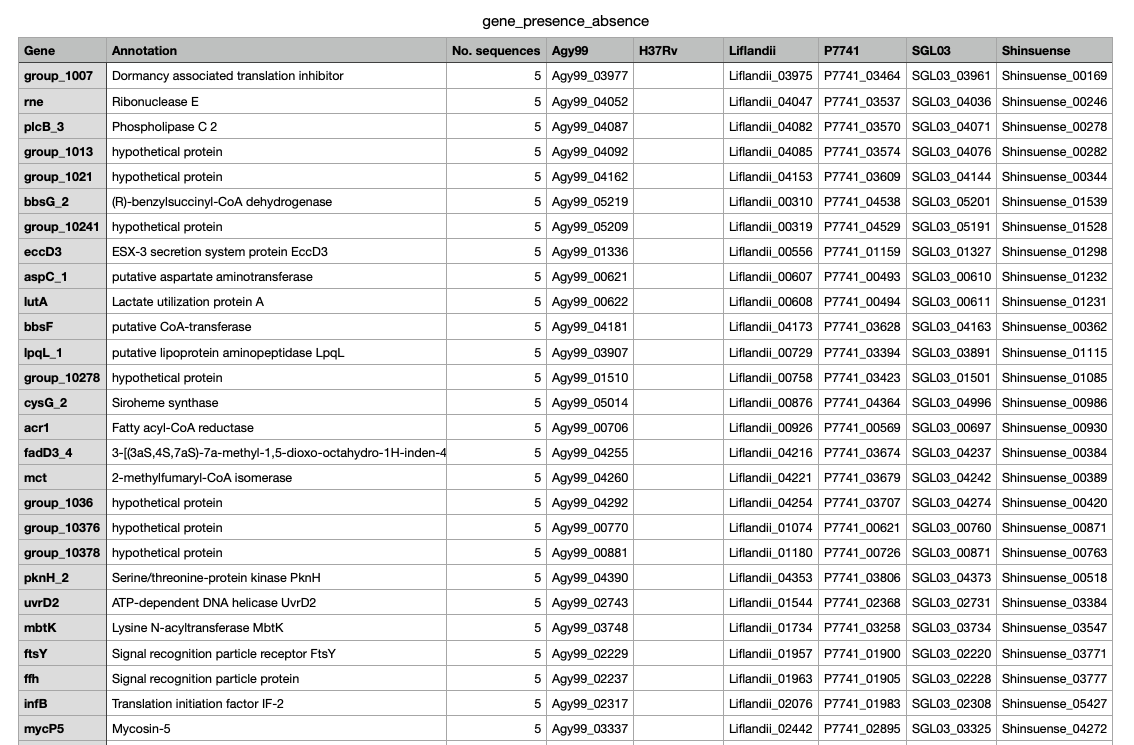
Image above: A sample of genes_presence_absense.csv where number of present = 5 genomes. Click on image to get the full CSV file - 11936 lines.
Image above: Pangenome Tree and Matrix of the Mycobacterium tuberculosis genome versus the Mycobacterium ulcerans genomes. Of the 11935 only 91 are considered core genes across all 6 genomes (a score of between >= 99% similarity). A click on the image gets the scalable SVG version.
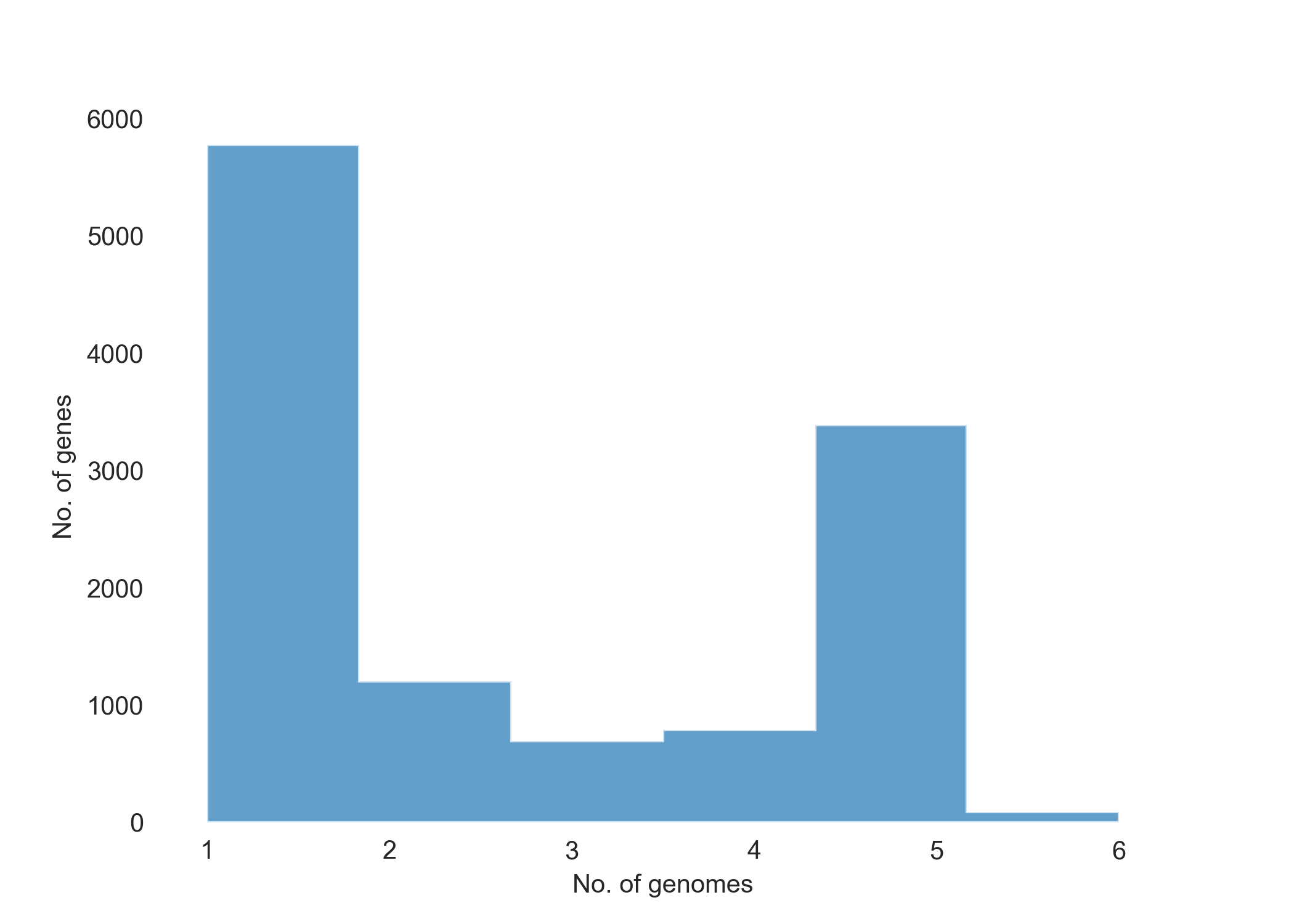
Pangenome Frequency.
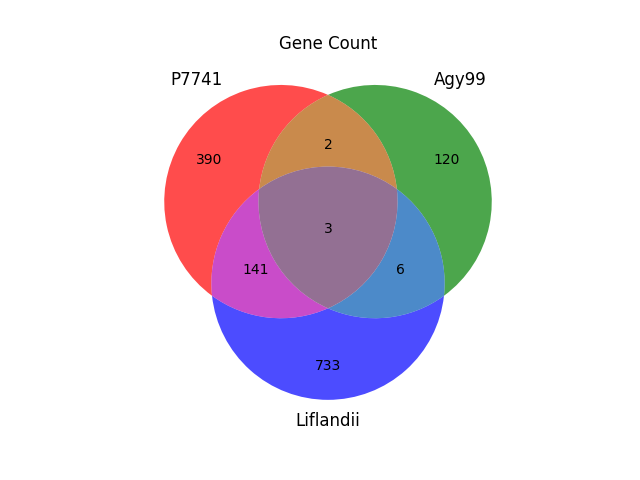
Genes unique to the set P7741, Agy99 and Liflandii (1395 = 390 + 2 + 120 + 141 + 3 + 6 + 733). Within the set, for example Agy99 has 120 unique genes. Of the 1395 unique genes within the set, Agy99 and P7741 share only 5, whilst P7741 and Liflandii share 144. Seems like P7741 and Liflandii are phylogenetically closer than to each other than to Agy99.
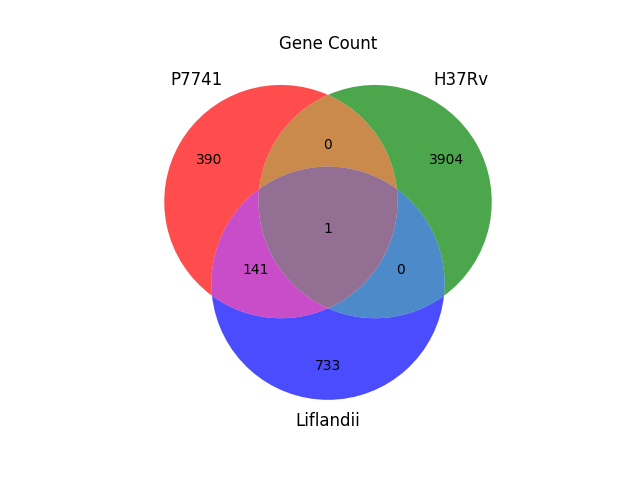
Genes unique to the set P7741, H37Rv, Liflandii (5169 = 390 + 0 + 3904 + 141 + 1 + 0 + 733). Mycobacterium tuberculosis (var. H37Rv) is clearly, and as expected, relatively distant to the Mycobacterium ulcerans genomes.
Bug note
# With Quarto, the strange bug means that if an {r} executable block is included# then the preceding {zsh} or {bash} blocks are executed, otherwise not!# Simply include this and all is executed as desired.