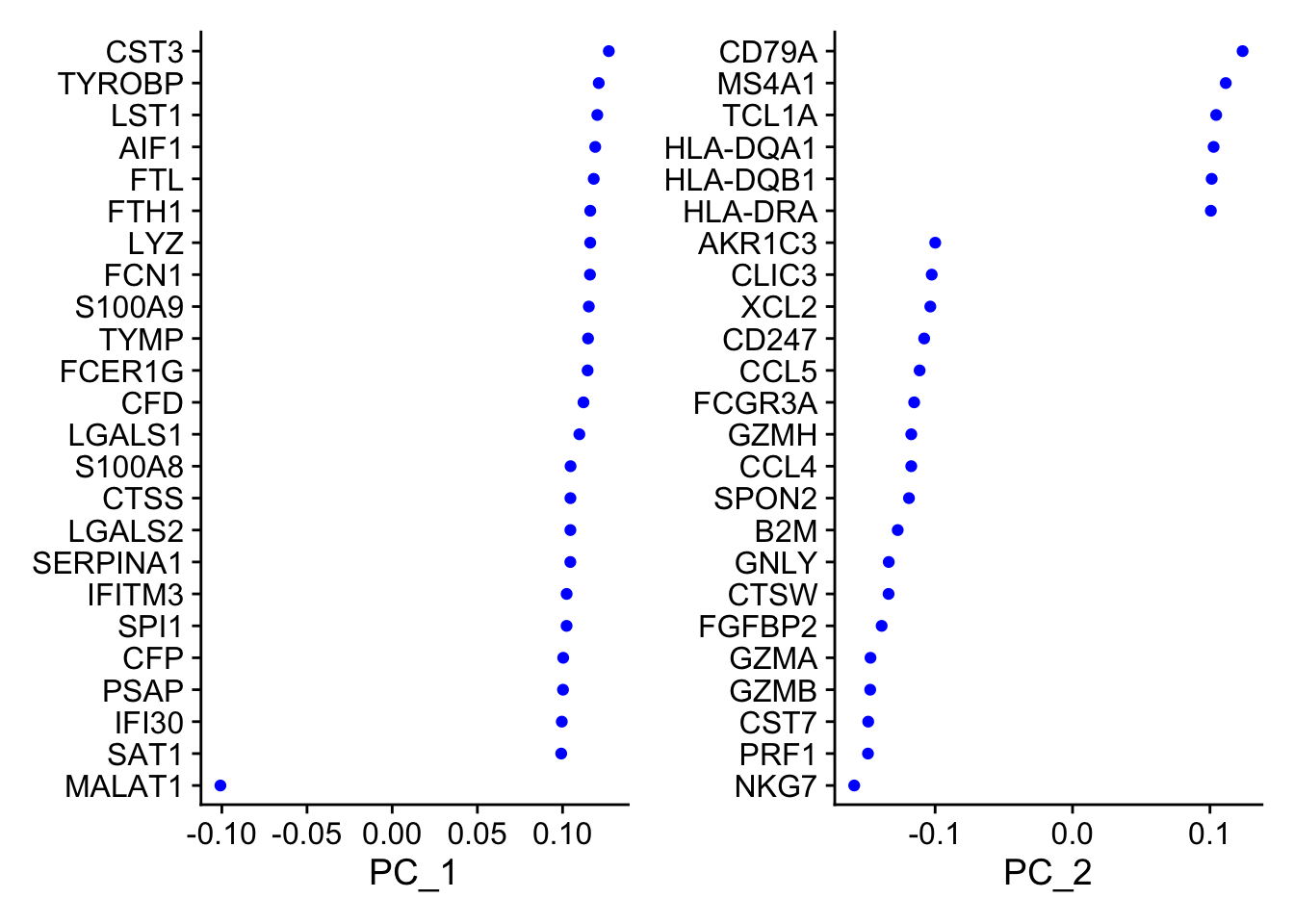
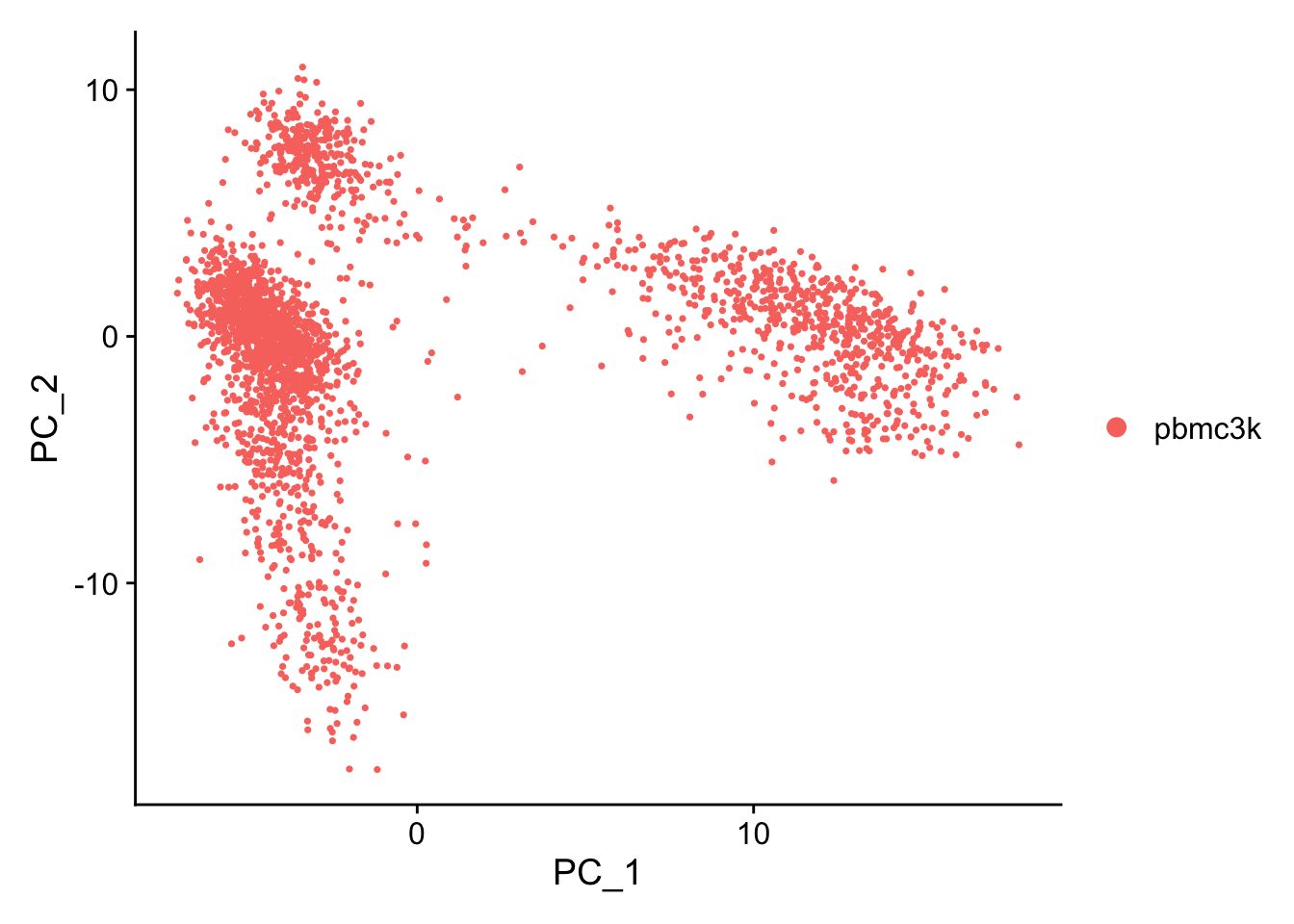
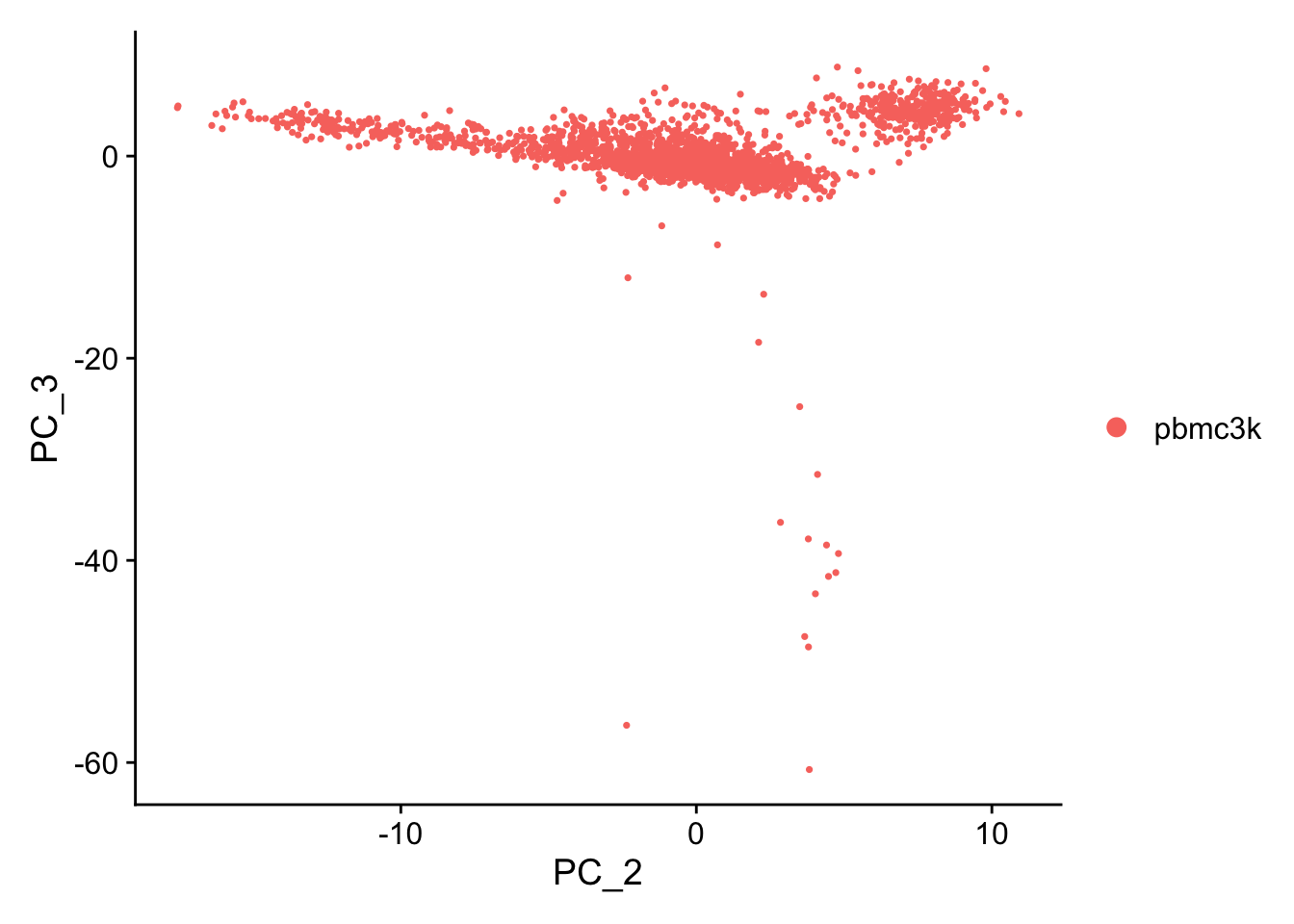
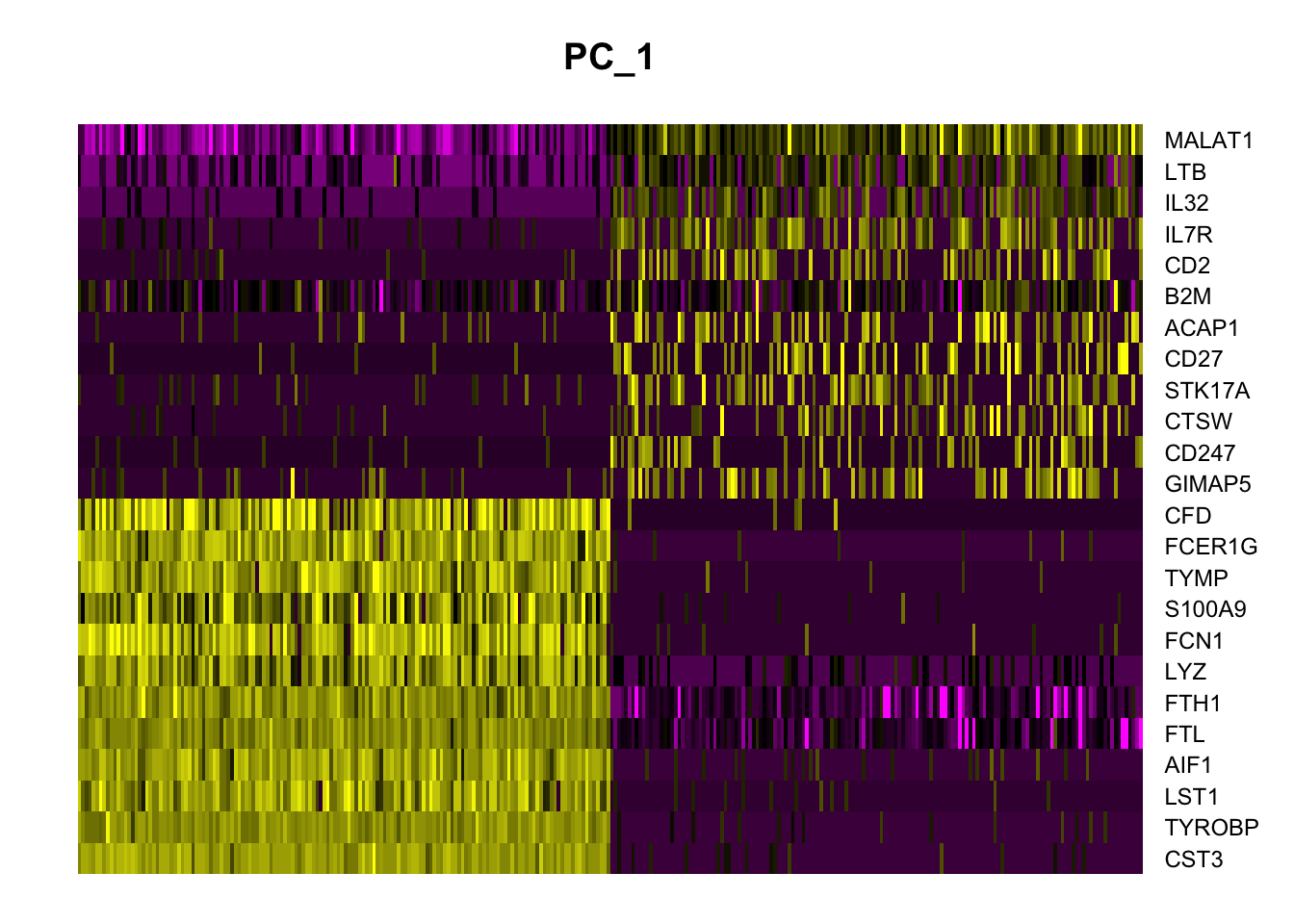
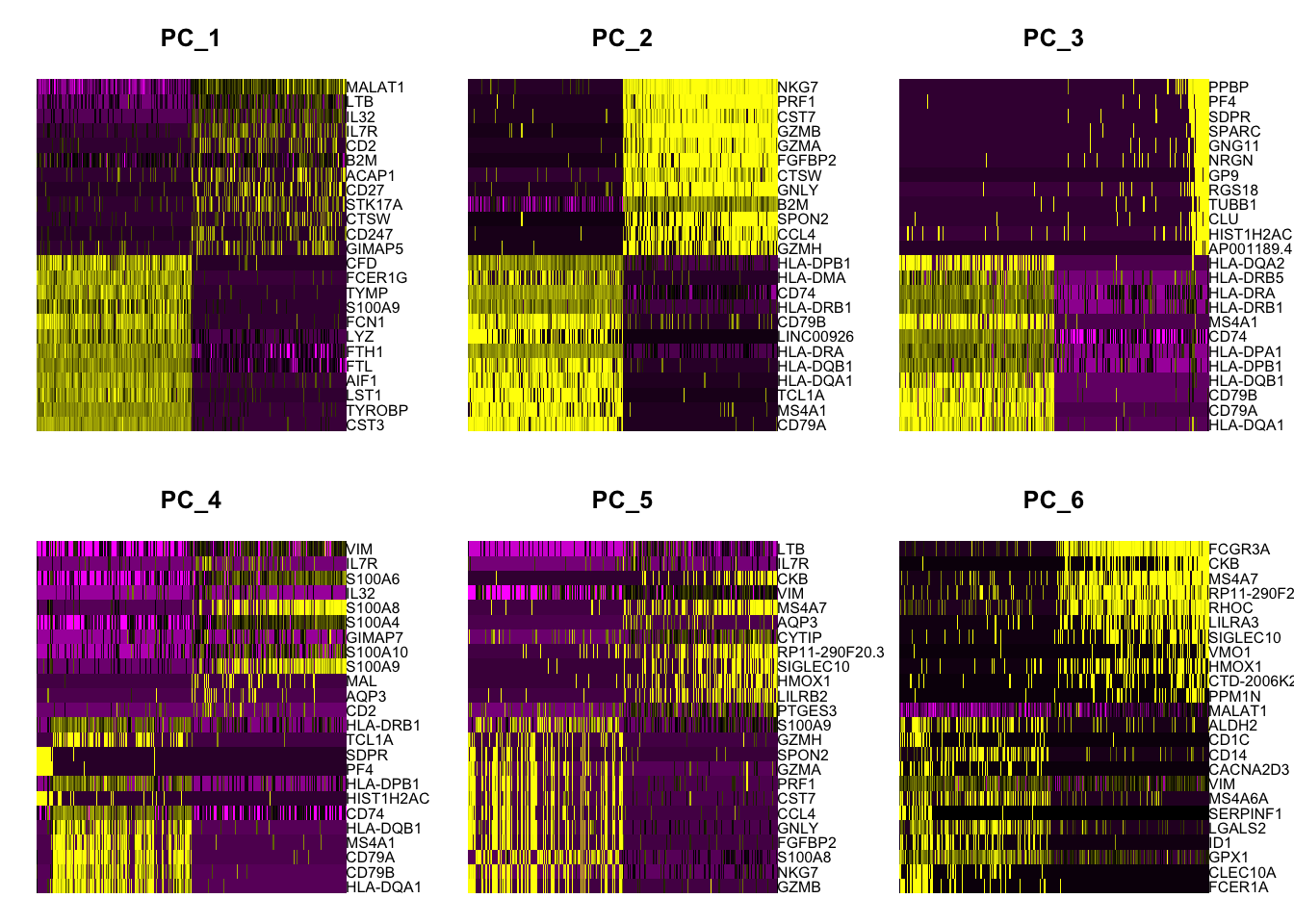
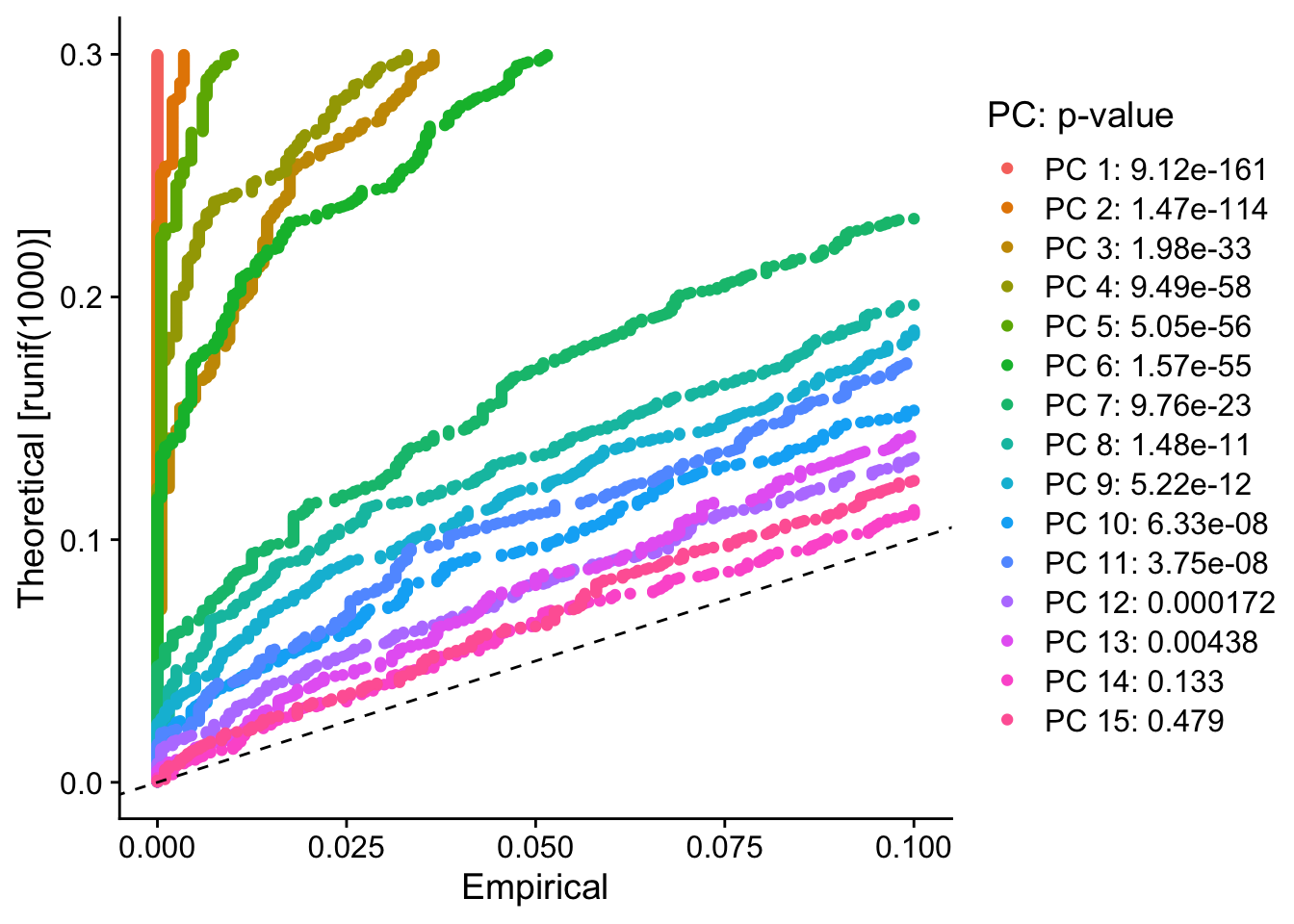
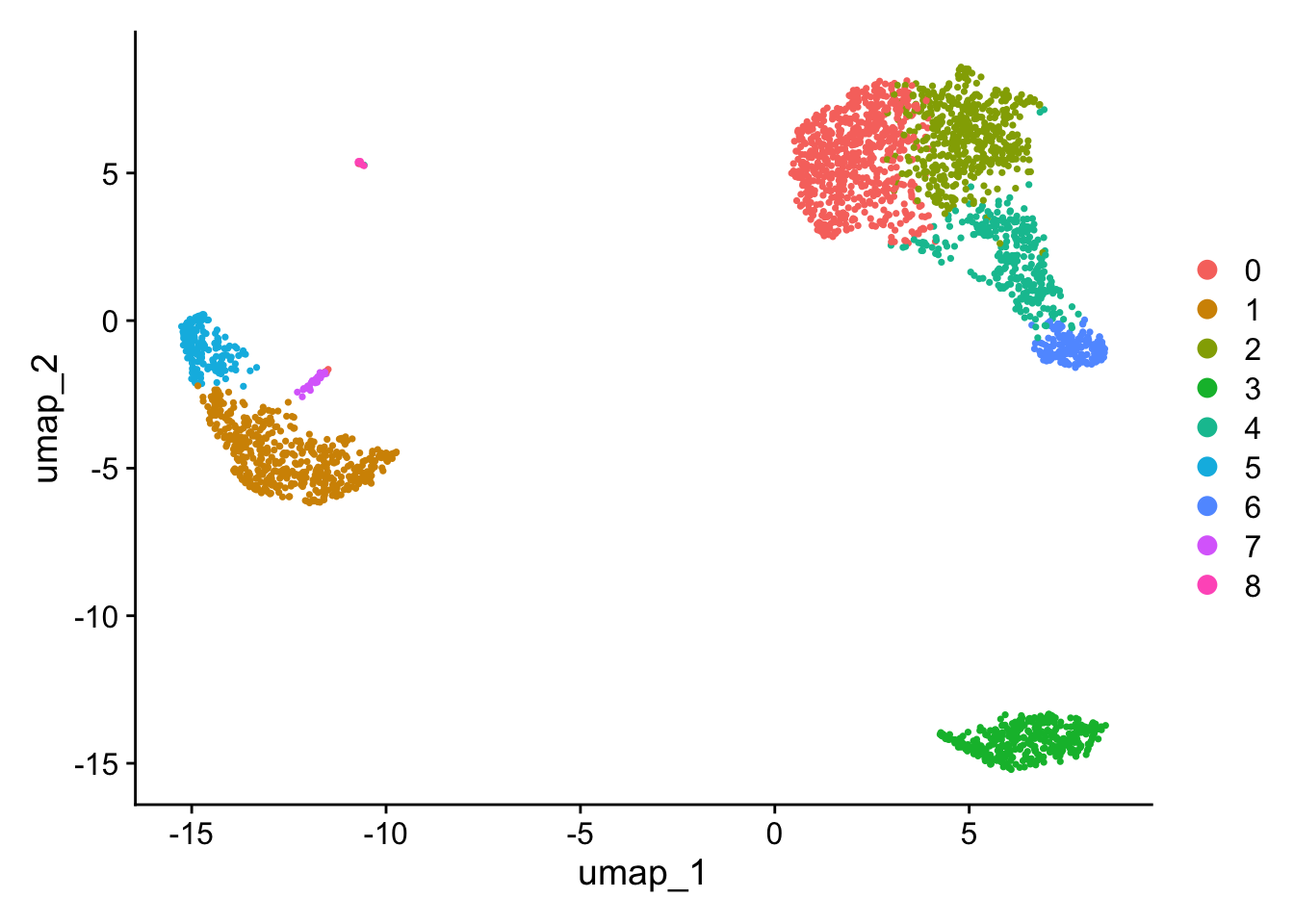
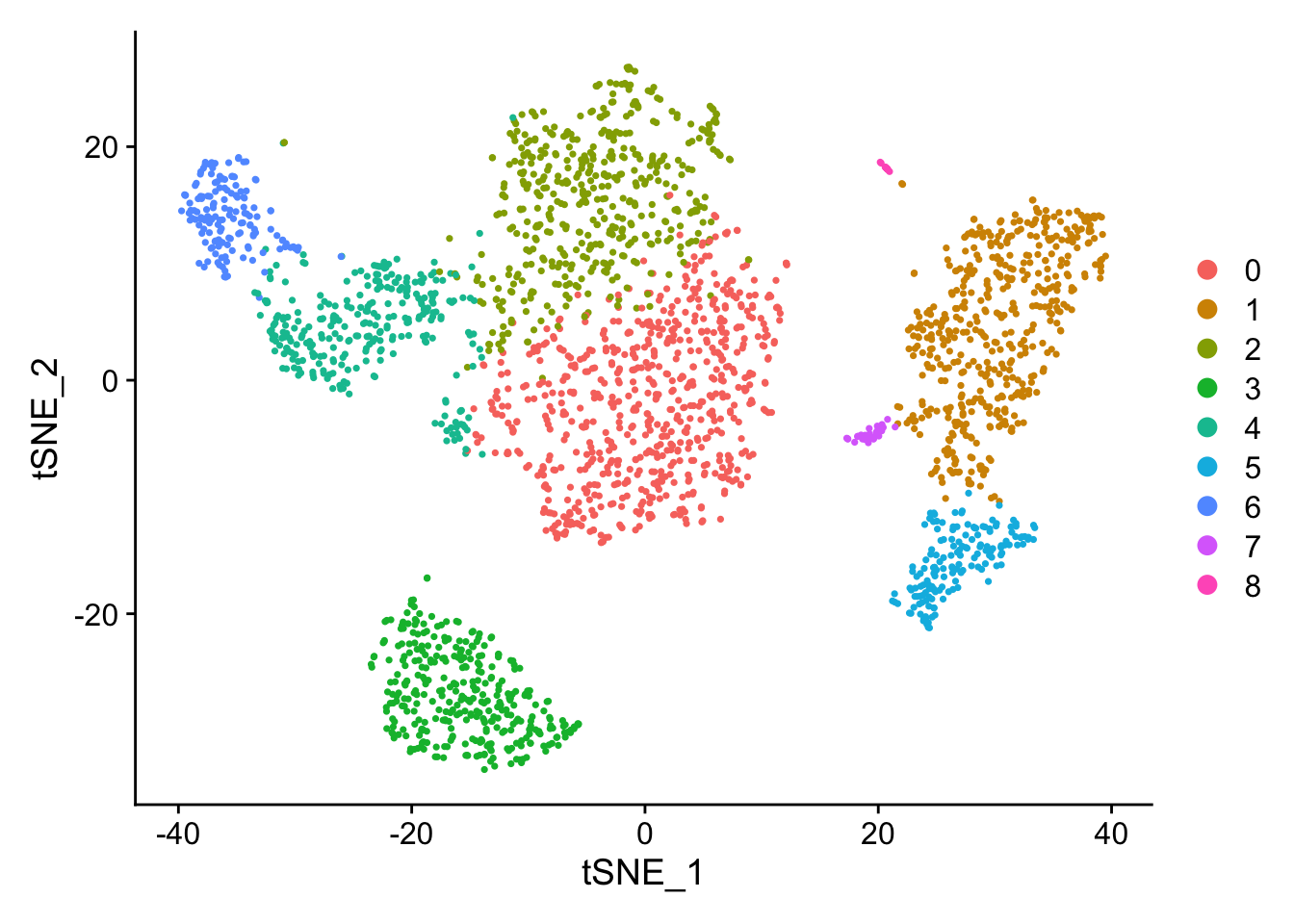
PC_ 1
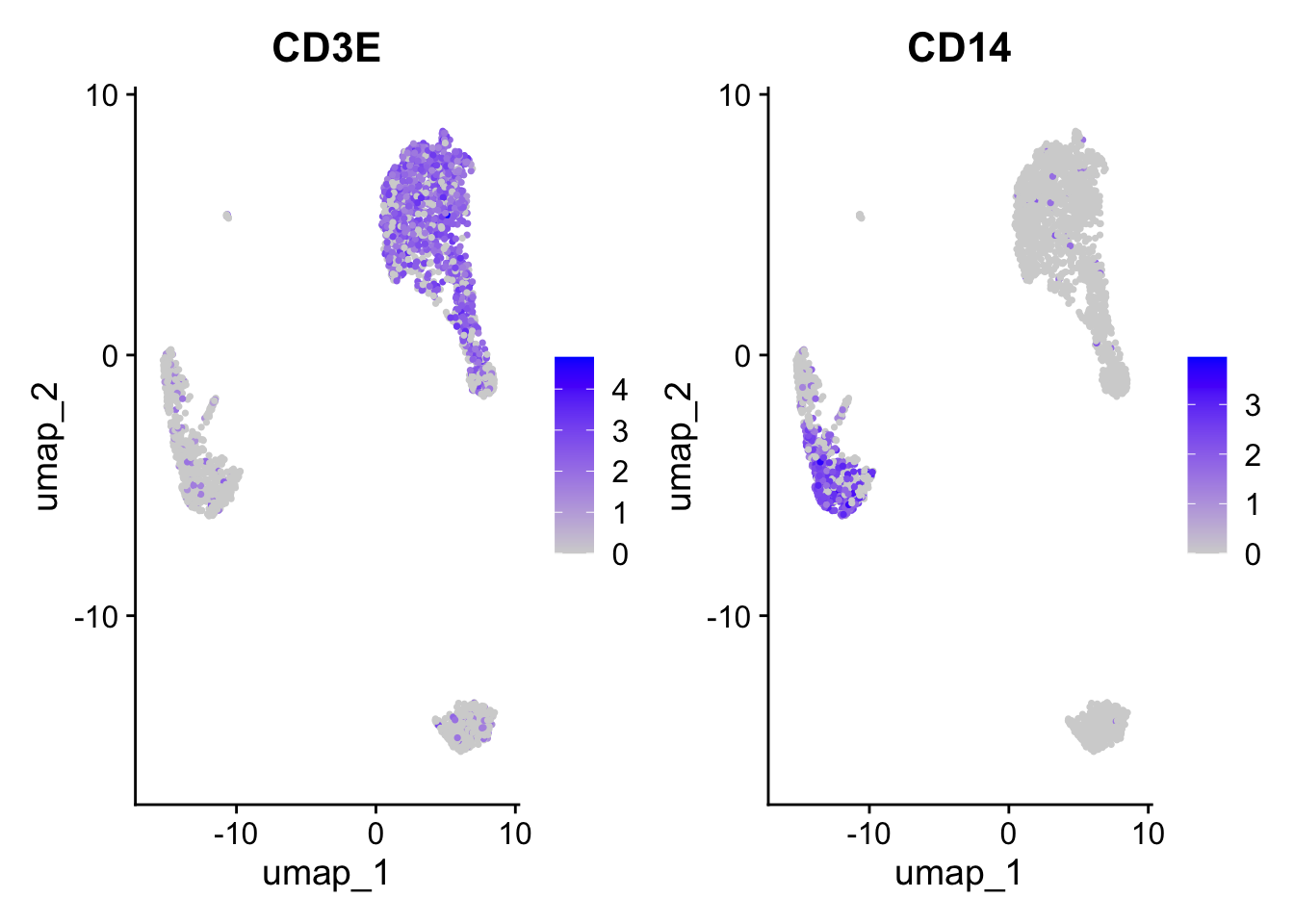
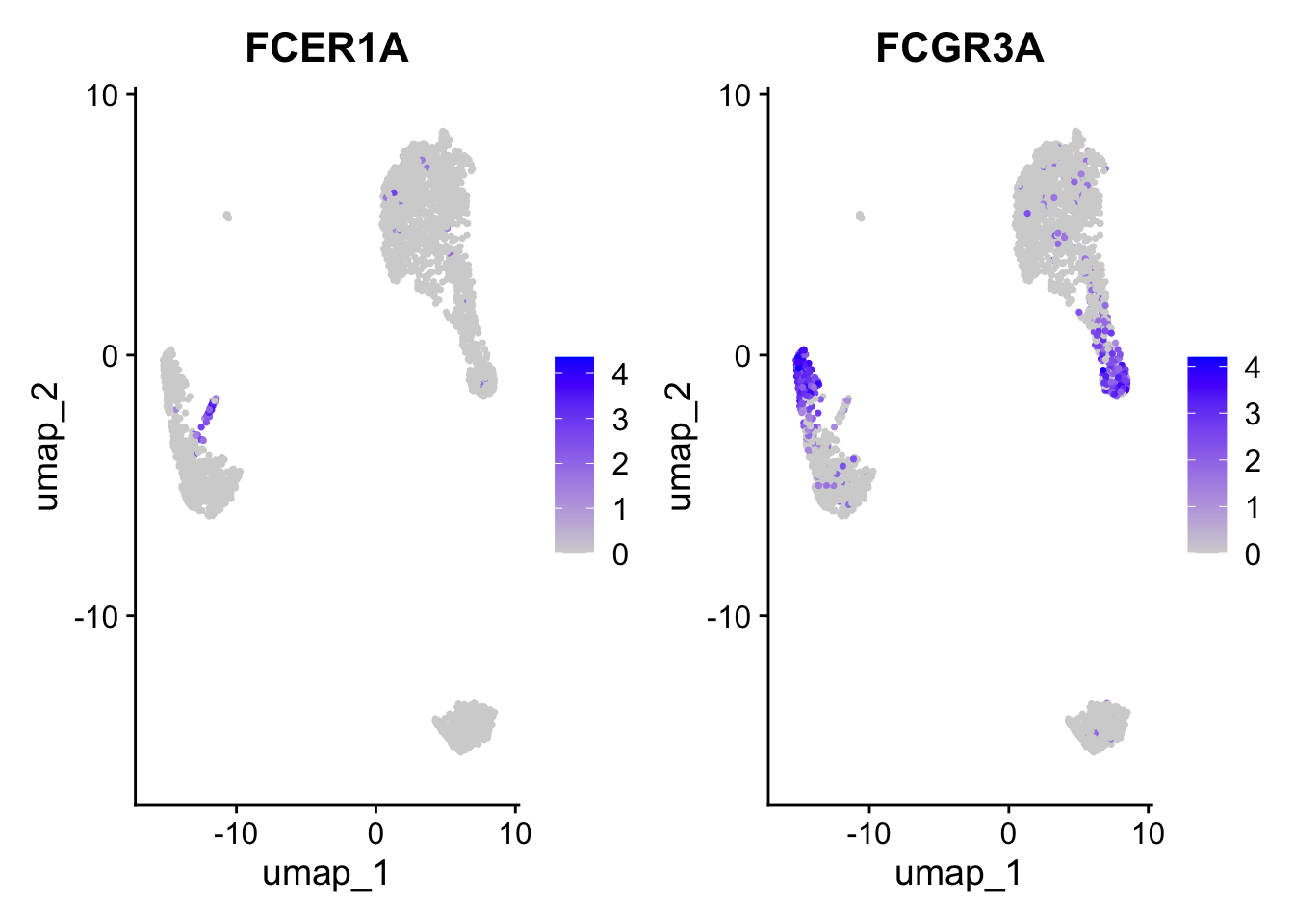
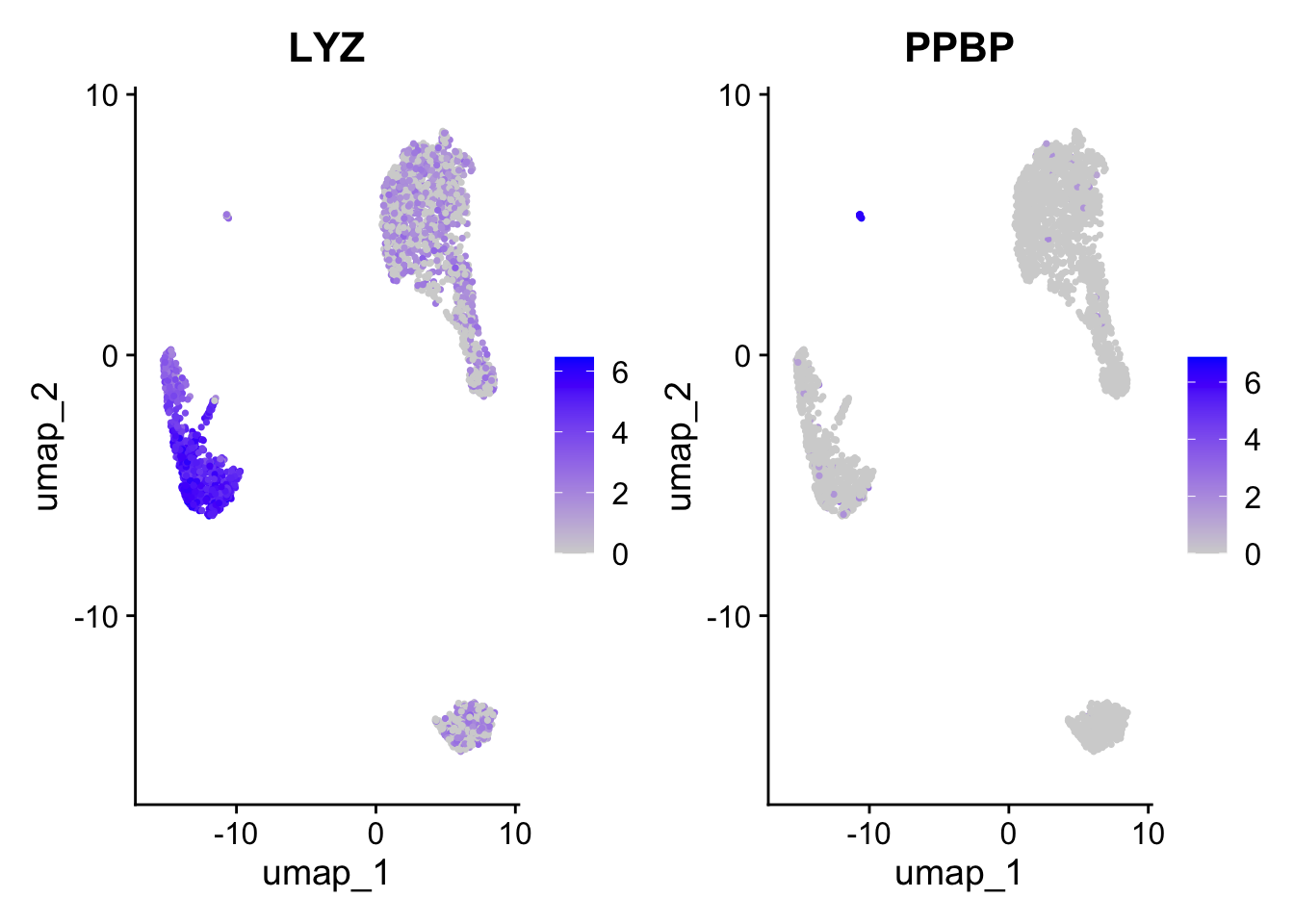
Positive: CST3, TYROBP, LST1, AIF1, FTL, FTH1, LYZ, FCN1
Negative: MALAT1, LTB, IL32, IL7R, CD2, B2M, ACAP1, CD27
PC_ 2
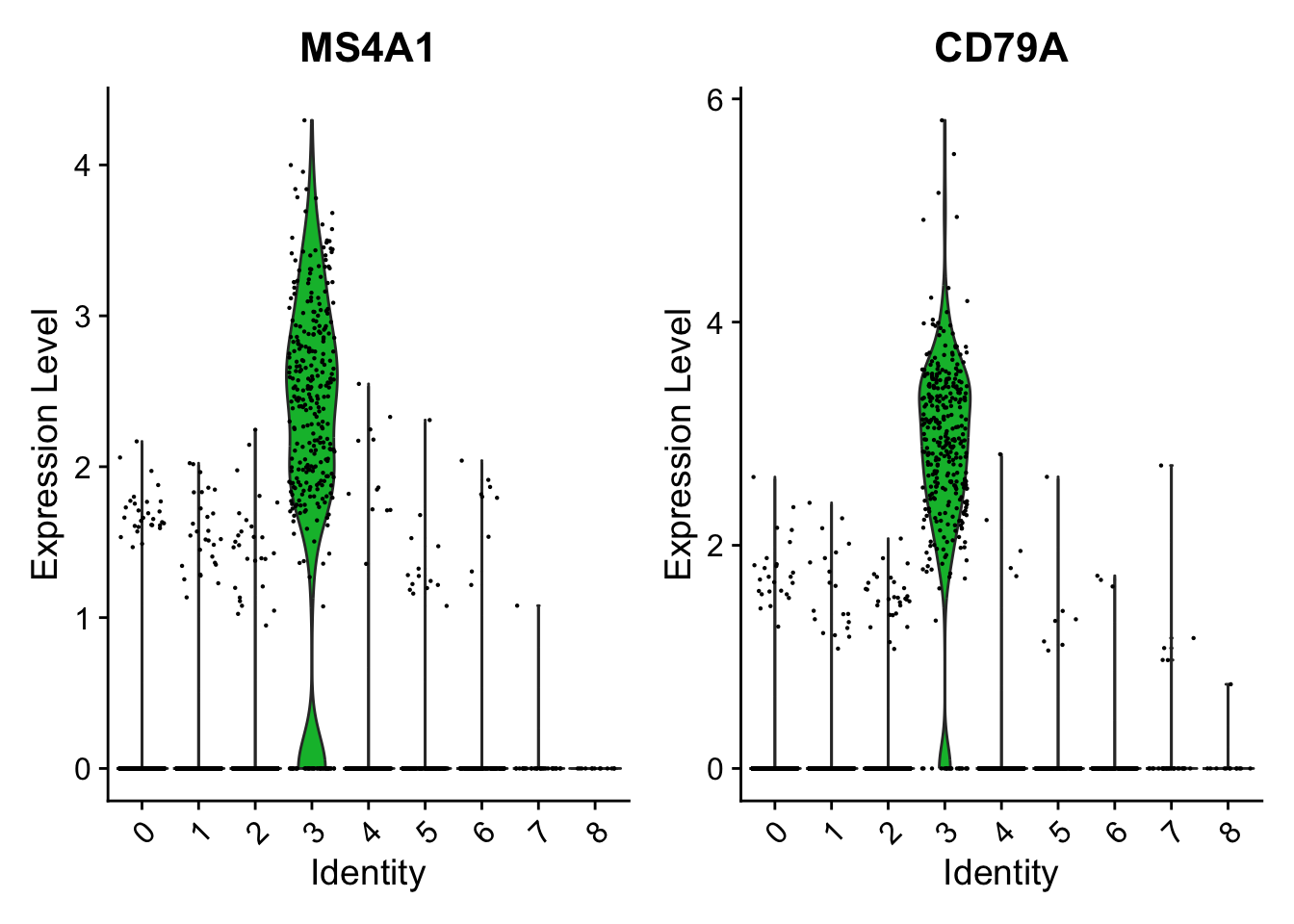
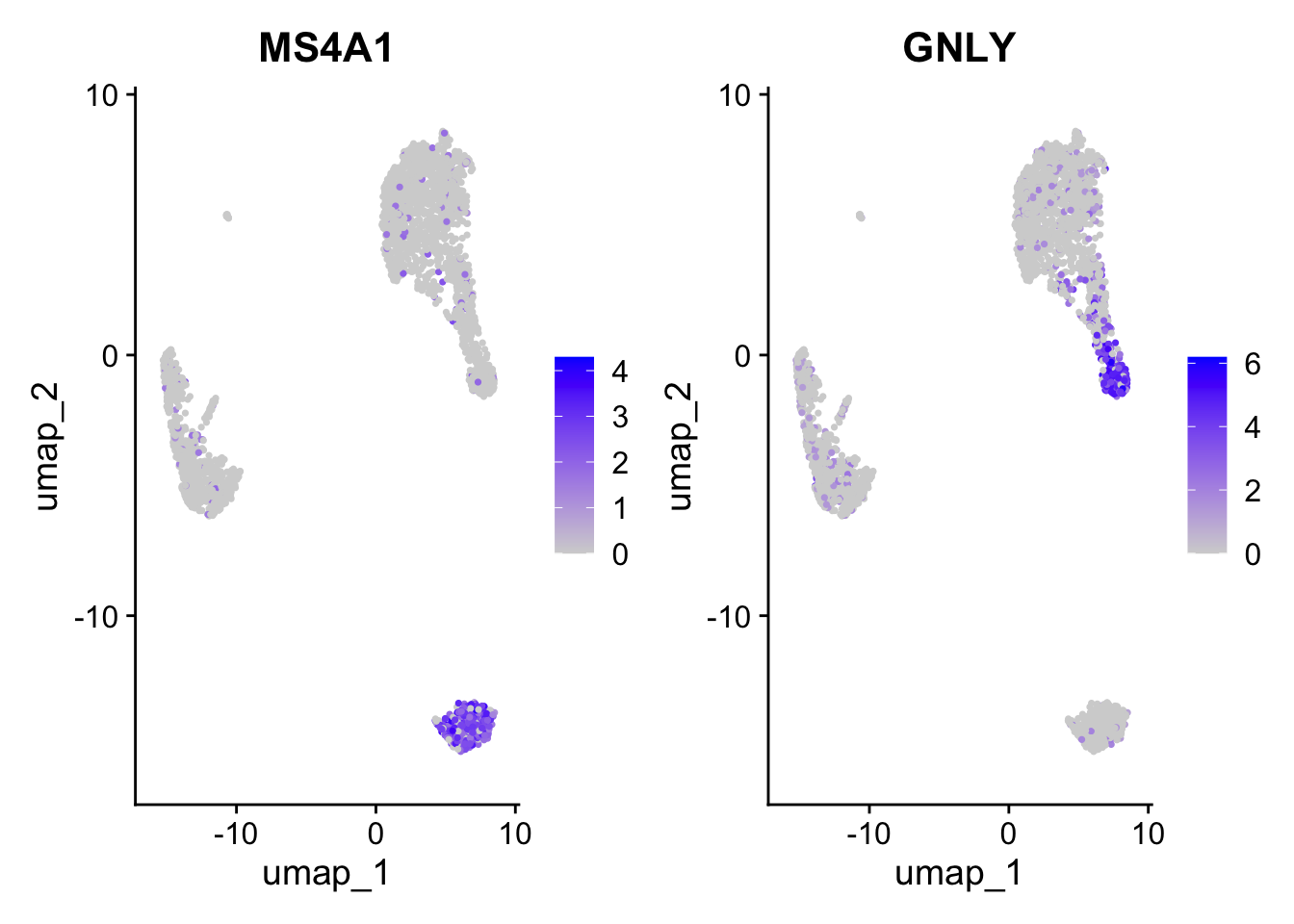
Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1, HLA-DRA, LINC00926, CD79B
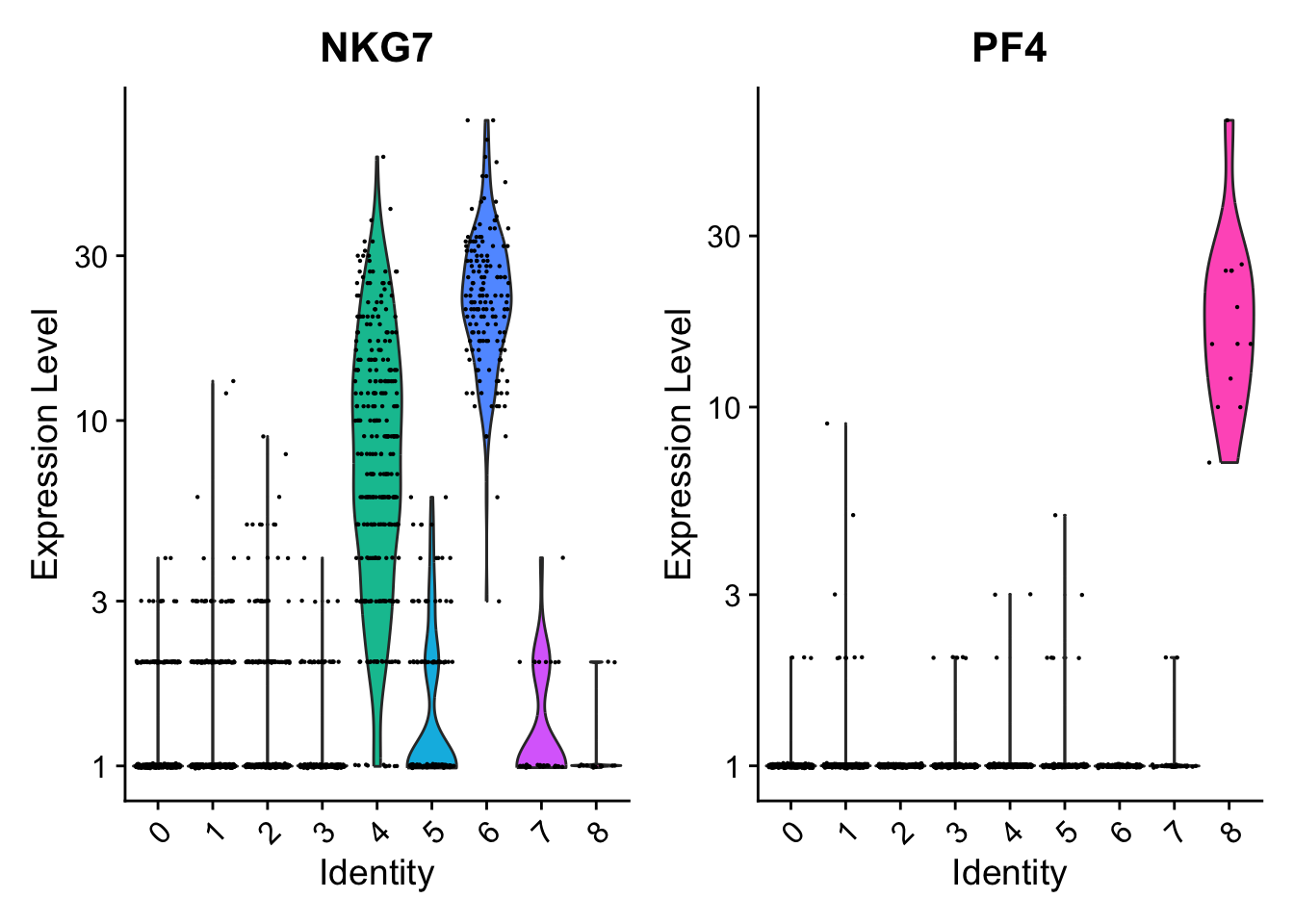
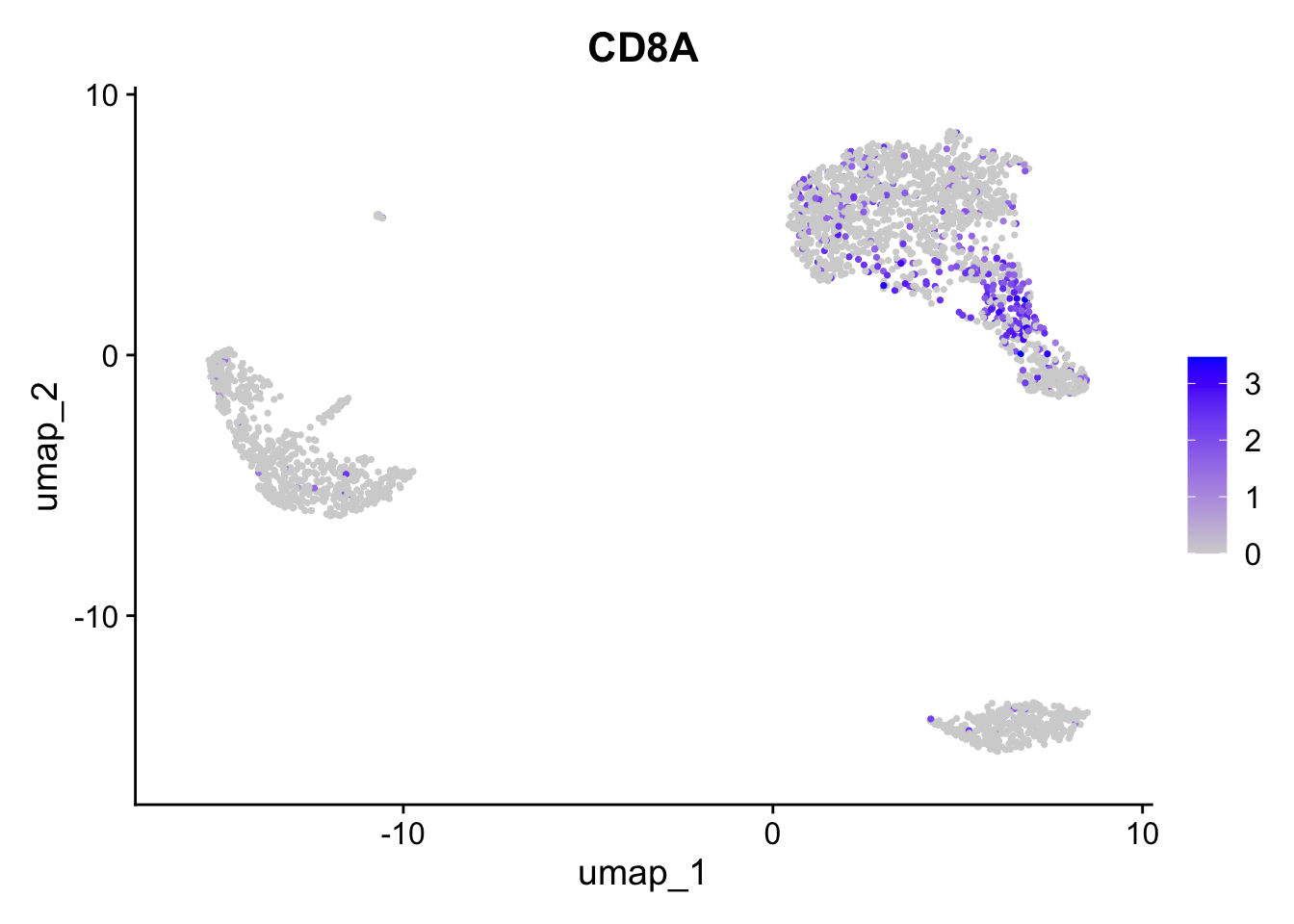
Negative: NKG7, PRF1, CST7, GZMB, GZMA, FGFBP2, CTSW, GNLY
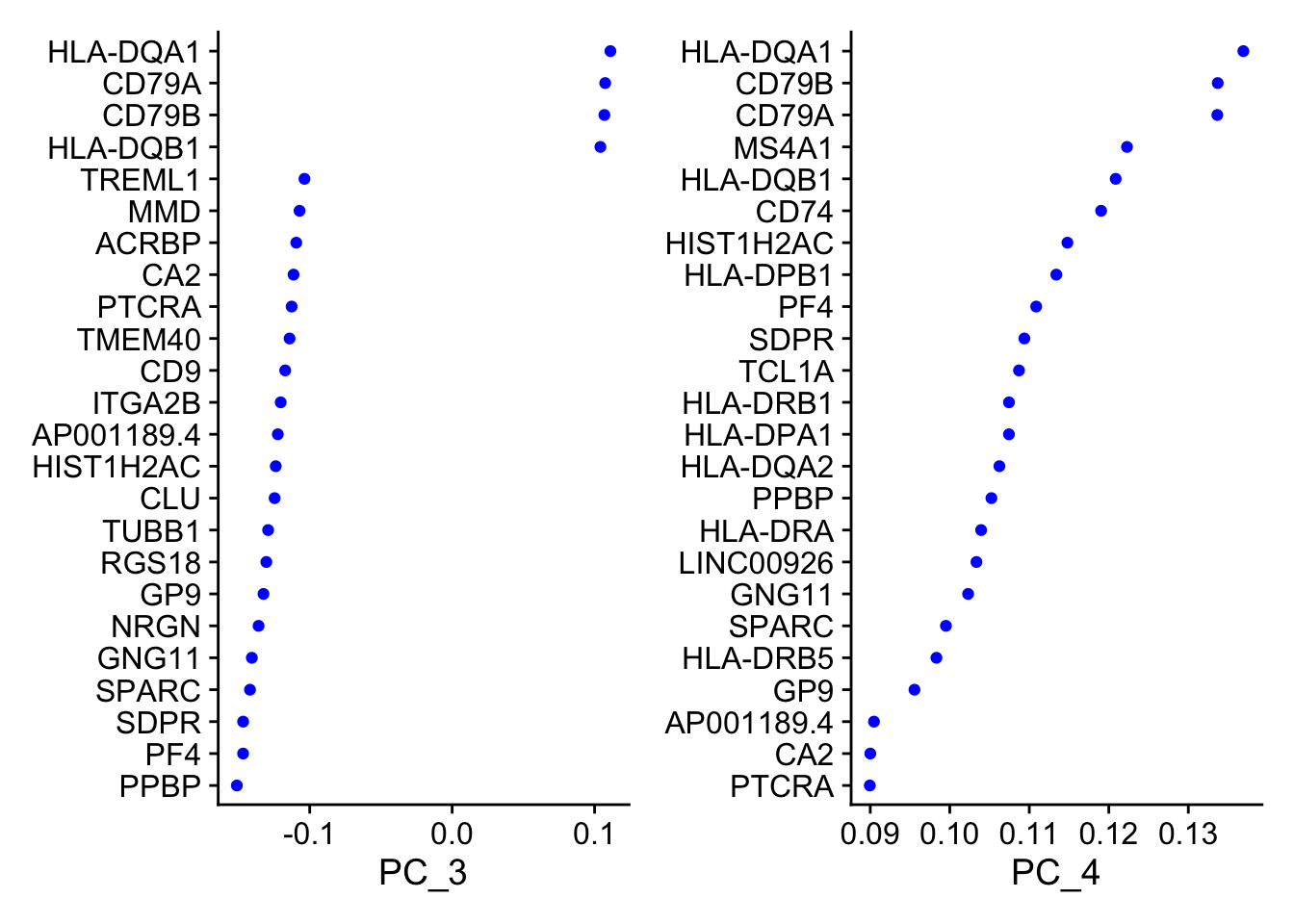
PC_ 3
Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1, HLA-DPA1, CD74, MS4A1
Negative: PPBP, PF4, SDPR, SPARC, GNG11, NRGN, GP9, RGS18
PC_ 4
Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1, CD74, HIST1H2AC, HLA-DPB1
Negative: VIM, IL7R, S100A6, IL32, S100A8, S100A4, GIMAP7, S100A10